
In the early days of forensic genetic genealogy, microarrays were considered for use in forensic testing. This was largely because microarrays had been widely used in the adjacent industry of consumer genetic genealogy. Major genetic genealogy databases were composed of DNA profiles built using microarrays, making it logical to attempt to use the same technology for forensics. However, unlike the high-quality DNA typically used in consumer applications, forensic DNA is often low quality and low quantity. This difference makes microarrays a terrible tool for forensics, as microarrays often fail to provide reliable data (or any data at all) when applied to forensic samples, especially those exhibiting degradation and DNA damage. There are many other disadvantages to using microarrays in forensics, but this is a topic for another post.
Why Microarrays Fall Short for Forensic DNA
Microarrays are sensitive to DNA quality and quantity because of the need for substantial quantities of intact DNA for hybridization—the process in which DNA fragments bind to complementary sequences on the microarray chip. When the DNA is degraded or scarce, hybridization efficiency is compromised, leading to low-quality or incomplete data. In the last several years, the field has recognized that building high-quality SNP profiles for forensic cases requires a method that can handle low-input and degraded DNA, something massively parallel sequencing (MPS) platforms, like Illumina’s, do exceedingly well.
Call Rate and Microarrays: A Link to Data Quality
Let’s look at why call rate is so closely linked to data quality in microarrays. Microarrays work by hybridizing fragmented DNA to pre-selected probes on a microarray chip surface, labeling the hybridized probes with fluorescent molecules, and then measuring the fluorescence emitted when the hybridized DNA is excited. Two key parameters determine whether a genotype can be successfully called for a specific SNP:
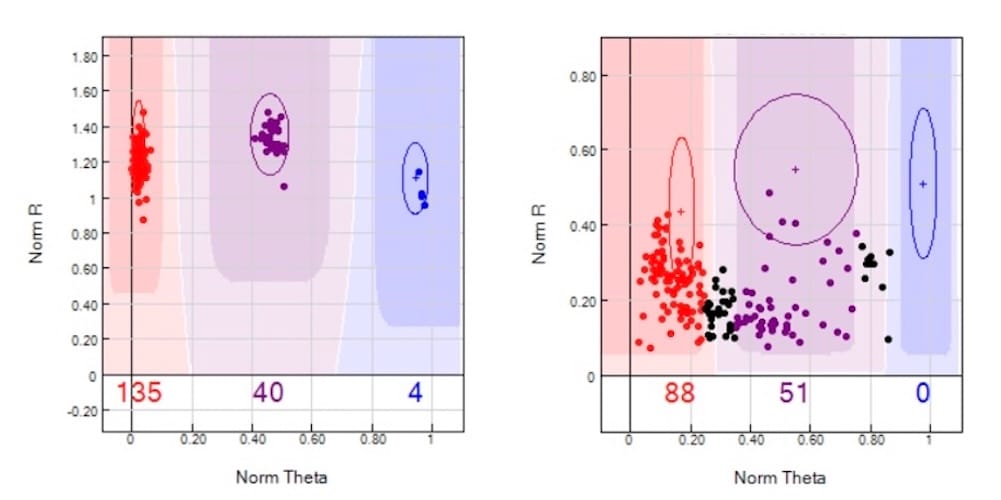
- Norm R: This is the normalized R value, representing the intensity of the fluorescence emission for a given SNP. If the intensity is too low, it may indicate that there is not enough DNA or that hybridization was inefficient, leading to a missed call. Conversely, if the intensity is too high, it could reflect poor quality DNA or technical artifacts.
- Norm Theta: This is a representation of the allele frequency of the SNP on a scale from 0 to 1. Ideally, homozygous AA would be represented as 0, heterozygous AB as 0.5, and homozygous BB as 1.0. If the DNA sample quality is low, the data may not cluster cleanly into these categories on the Theta axis, leading to ambiguous results and failed genotype calls.
Take a look at the image below. You can see in the left panel microarray data clustering as expected, which will lead to genotype calls. When the two parameters—norm R and norm Theta—fall outside their expected ranges, the likelihood of making an accurate call drops significantly. This is why call rate is such a reliable indicator of data quality in microarrays.
Poor quality DNA will produce noisy signals, causing both the intensity (R) and allele frequency (Theta) data to fall outside expected ranges or thresholds, which in turn results in a low call rate (see the right panel, in the image below). As an additional note, it is worth mentioning that DNA mixtures, or DNA samples for which there are more than one contributor, will almost certainly throw off norm Theta. This yet another reason why it is very dangerous to use microarrays for forensic analysis – especially in sexual assault cases.
Call Rate in Sequencing: A Misleading Metric
When it comes to DNA sequencing, call rate is a far less informative measure of data quality. Massively parallel sequencing platforms, like Illumina’s Sequencing by Synthesis (SBS) approach, read DNA in much greater depth and breadth, making call rate less relevant.
In sequencing, call rate simply reflects the proportion of positions in the genome where a base has been called. However, this doesn’t provide any insight into the quality of those calls. A high call rate in sequencing might still be accompanied by poor data quality if the sequencing depth is too low or if there are errors in base calling. In forensic genetic genealogy, where degraded or low-input samples are the norm, a high call rate may falsely suggest that the data is of good quality, when in fact, it may not be reliable enough to draw meaningful genetic matches.
Instead of relying on call rate, forensic genetic genealogists should prioritize metrics like sequencing depth—the number of times each base is read during the sequencing process. This is particularly important for detecting heterozygous variants, which are key to accurately determining identity-by-descent (IBD) segments. IBD segments, which reveal shared ancestry, are often terminated by homozygous calls. If heterozygous variants are missed due to insufficient sequencing depth, the resulting genetic matches can be incomplete or inaccurate.
Conclusion: Choose the Right Tools & Metrics
Call rate is a useful quality control metric for microarrays, where targeting predefined SNPs is the goal, but don't get tricked into using it to evaluate DNA sequencing, where a more comprehensive and nuanced understanding of data quality is required.
At Othram, we were among the first to advocate for comprehensive DNA sequencing as the method of choice for building DNA profiles from forensic samples. We have also long promoted data quality metrics that extend beyond simple call rates. In forensic genetic genealogy, the goal is to create ultra-sensitive DNA profiles capable of detecting distant genetic relationships. Inadequate or incomplete DNA profiles can severely undermine the effectiveness of genetic genealogy, making it difficult or impossible to solve your case.
If you are not ready to onboard this new technology in your own forensic setting yet, come to Othram. Our team operates the world's first purpose-built forensic laboratory for forensic genetic genealogy. We developed Forensic-Grade Genome Sequencing® or FGGS® to enable ultra-sensitive detection of distant relationships. It's part of our Multi Dimensional Forensic Intelligence (MDFI) platform.
More forensic genetic genealogy cases have been solved with Othram FGGS® than any other method. Let’s work together to unlock answers and bring justice to those who need it most. Get started here.





