
Families are central to many areas of study, from sociology to anthropology, and even the natural sciences. In genetics—the science of inheritance across generations—the family is the core unit through which scientists study genetic relationships and inheritance. We intuitively understand that we are genetically closer to our parents than we are to our grandparents or cousins. Also it is well known that full siblings are more closely related than half-siblings. But how do we translate this intuitive notion of “closeness” into something quantitative? Fortunately, scientists have developed a mathematical framework to measure the full range of genetic relationships.
In the early 20th century, R.A. Fisher formalized a concept based on Mendelian genetics: each step in relatedness reduces genetic similarity by half. This concept means that individuals inherit 50% of their DNA from each parent, 25% from each grandparent, and 12.5% from each great-grandparent. The same logic applies laterally—an individual’s relatedness to an aunt or uncle is about 25% because of sharing a relationship with two grandparents. This principle of genetic dilution forms the foundation of our understanding of genetic relationships within a family tree.
From Theory to Practice: How Relationships Were First Traced
While this theoretical framework is clear, measuring real genetic relationships is more nuanced. In the 1960s, scientists began to trace relationships using molecular markers, and by the 2000s, they were able to assess these relationships more effectively, scanning across the three billion base pairs of our genome.
Before the human genome draft was completed in 2001, geneticists relied on simpler biochemical markers to estimate relatedness. One widely used method was blood group analysis, where scientists examined the inheritance of A, B, AB and O blood types. These blood groups are determined by specific antigens, and their inheritance follows Mendelian rules. For instance, because the O allele is recessive, two O-type parents cannot have an A, B, or AB child. While useful, blood group analysis had limitations, particularly the phenotype blood group A could not be distinguished as being genetically AA or AO and similarly blood group B could be either BB or BO.
The Rise of STR Markers in Forensics
In the late 1980s, blood group analysis and other protein marker systems were replaced by a more advanced technique: DNA typing. In the 1990s STR (short tandem repeat) markers became established and today still are the primary genetic systems analyzed by the forensic genetics community. STRs are regions of the genome that mutate rapidly and generate substantial variation. Concomitantly with research and development of STR testing , the DNA Identification Act of 1994 officially authorized the establishment of a national DNA database in the United States. This act laid the legal framework for the creation of the National DNA Index System (NDIS), which became operational in 1998. NDIS is one part of the FBI's Combined DNA Index System (CODIS).
For siblings, who share about 50% of their genetic material, the pattern of shared alleles differs due to the random mix of genetic markers inherited from both parents. STR markers are highly variable, which makes it easy to match close relatives like parents and children, as they share at least one allele at each marker. However, the STR profiles, that populate CODIS, are ineffective at measuring genetic relationships beyond parent/child or full siblings. Even for half-siblings, the variability and the small number of STR markers in a CODIS profile make it unsuitable for assessing the genetic relationship. This limitation becomes even more pronounced when trying to identify more distant relationships, like cousins, as the STR systems currently in CODIS were not designed for those relationships.
From STRs to SNPs: The Next Leap in Genetic Analysis
The early 2000s marked a major breakthrough in genetic analysis with the introduction of genome-wide microarrays. Illumina’s release of a platform capable of analyzing 650,000 single nucleotide polymorphisms (SNPs) revolutionized genomics, significantly increasing the number of genetic markers available for analysis. While an individual SNP carries less information than an individual STR—most SNPs have only two alleles while generally STRs have from 5 to 30 alleles—the sheer volume of SNPs measured provides far more genetic data, allowing for a much finer and more granular analysis of genetic variation.
In contrast, STR markers (as used in systems like CODIS) could only analyze variation at a limited number of markers, typically 20 to less than 30. SNP arrays, on the other hand, enable scientists to analyze hundreds of thousands, or even millions, of positions across the genome. This volume of data dramatically improved the ability to detect not just close relationships like parent/child or full siblings, but also more distant relationships like half-siblings, cousins, grandparents, and beyond.
However, genetic similarity between relatives doesn’t arise uniformly. For example, although both grandparents and aunts/uncles share approximately 25% of their DNA with a person, the pattern of inheritance is different. Grandparents pass down DNA in two stages—from grandparent to parent to grandchild—while aunts and uncles share two common grandparents with the individual’s parents. Understanding these subtle differences is crucial for forensic genetic analysis, where detailed patterns of shared DNA segments are used to determine how closely two individuals are related.
Dense SNP Testing Enables Segment Matching
With dense SNP data, segment-based matching has become one of the most reliable ways to measure genetic relationships. But what exactly is a “segment,” and how is it used to trace connections between individuals?
A segment refers to a continuous stretch of DNA inherited from a common ancestor. When DNA is passed from parents to children, it is passed in large segments. These segments consist of multiple genetic markers (such as SNPs) that are transmitted together from one generation to the next.As DNA is inherited across multiple generations, these segments get broken into smaller pieces through a process called recombination.
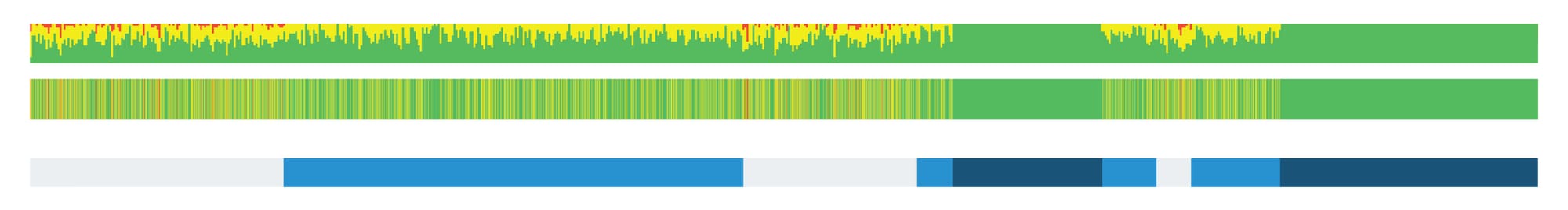
To compute a segment, scientists look at SNP data to find stretches of DNA that are identical between two individuals. A segment is identified when these shared SNPs and their genetic states (i.e., A, G, C or T) are in the same sequence and order across a long enough stretch of the genome. The length of a shared segment is typically measured in centimorgans (cM), a unit that reflects the likelihood of recombination events breaking up that segment over generations and is somewhat similar to, but not exactly, the physical distance along the chromosome.
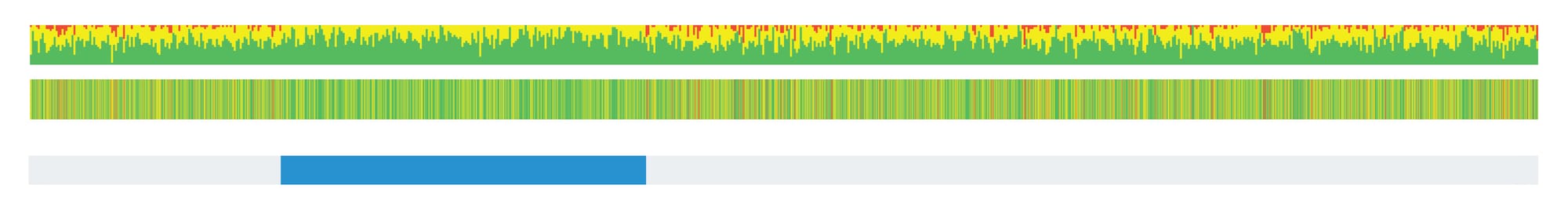
When analyzing relationships using segment data, two key measurements help determine genetic relatedness:
- Length of the Largest Shared Segment: The longer the largest segment of shared DNA between two individuals is, the closer their genetic relationship is likely to be. For example, parents and children will share long segments of DNA, while more distant relatives (such as cousins) will share shorter segments.
- Total Amount of Shared DNA Across All Segments: In addition to looking at the longest shared segment, analysts also consider the total amount of DNA shared across all segments. This measurement provides a more complete picture of the genetic relatedness between two individuals. Close relatives will share more total DNA, while distant relatives will share less.
Both of these measurements—largest segment length and total shared DNA—are crucial for determining how closely two people are related. Together, they allow geneticists to more accurately assess relationships between individuals based on the segments of DNA they inherited from common ancestors.
Sparse SNP Data: What to Do When You Can’t Measure Segments
While segment-based matching is a powerful tool when working with dense SNP data, it becomes much harder to use when SNP data are sparse or incomplete. If there are not enough SNPs to confidently identify long, shared segments, other methods can be used to infer genetic relationships.
In cases where segment matching is not possible, researchers can turn to statistical methods like kinship coefficients. These coefficients measure the overall similarity of genetic data between two individuals. Correlation-based methods can still provide valuable insights into genetic relationships, especially when working with sparse or incomplete datasets, but are less effective for detecting distant relationships.
However, relying on sparse SNP data has further limitations beyond just the loss of precision. Many advanced genetic genealogy tools, such as triangulation, rely on dense SNP data to identify shared segments across multiple individuals. Triangulation is crucial for confirming common ancestry among three or more individuals by ensuring they share identical segments of DNA in the same location on the genome. Without dense SNP data, tools like triangulation cannot be applied, limiting the analysis to simpler pairwise comparisons. Limited SNP data can be a major drawback when trying to map out complex family trees or to investigate distant relationships across multiple individuals.
For robust forensic and genealogical analysis beyond just comparing relationships between two people, dense SNP data are essential to take full advantage of advanced tools like triangulation, segment comparison, and other techniques designed to explore more intricate family connections.
Forensic Genetic Genealogy in Action
Over the last 50 years, forensic genetics has advanced from crude blood group analysis to a near-total genomic characterization for human relatedness. Modern genomic data, combined with decades of theoretical and practical work, have transformed our ability to trace familial relationships, bringing both science and technology together to deliver results that were once unimaginable.
The power of this technology was on full display with the resolution of Carla Walker’s case, a horrific crime that went unsolved for nearly half a century. In 2020, advanced DNA technology and genealogical analysis finally identified her killer, Glen Samuel McCurley, bringing resolution to a case that had baffled investigators for decades. By combining decades of genetic theory with modern DNA sequencing and genealogical tools, investigators were able to trace the DNA from distant relatives to help identify the source of the probative crime scene evidence.
This breakthrough technology is not only solving cold cases but also delivering real-time results in active investigations. In the 2023 murder of Rachel Morin, investigators used forensic genetic genealogy to track down her killer, Victor Martinez-Hernandez, who was from El Salvador. Although not a resident of the United States, the basic principles of measuring genetic relationships through familial DNA allowed investigators to trace him to the crime scene evidence, highlighting the global reach of this technology. The rapid identification of the suspect demonstrated how forensic genetic genealogy has transformed the pace and scope of criminal investigations.
The Best Case Scenario for Your Case
If you are using forensic genetic genealogy in your forensic investigation, you will need ultra-sensitive profiles optimized for detecting distant genetic relationships. If you aren't aiming to detect all relatives, you are doing it wrong. Inadequate, incomplete, or inaccurate DNA profiles can severely compromise the effectiveness of downstream genetic genealogy, making it difficult or even impossible to resolve complex cases.
If you are not ready to onboard this new technology in your own forensic setting yet, come to Othram. Our team operates the world's first purpose-built forensic laboratory for forensic genetic genealogy. We developed Forensic-Grade Genome Sequencing® or FGGS® to enable ultra-sensitive detection of distant relationships. It's part of our Multi Dimensional Forensic Intelligence (MDFI) platform.
More forensic genetic genealogy cases have been solved with Othram FGGS® than any other method. Let’s work together to unlock answers and bring justice to those who need it most. Get started here.





